 A nine-year-old healthy girl at summer camp, running up a hill to chase her friend, collapses and fractures her left arm. She is seen in the local emergency department where she is diagnosed with a supracondylar fracture and dehydration. Three months later her brother is found dead in his bed in the morning.
A nine-year-old healthy girl at summer camp, running up a hill to chase her friend, collapses and fractures her left arm. She is seen in the local emergency department where she is diagnosed with a supracondylar fracture and dehydration. Three months later her brother is found dead in his bed in the morning.
Are emergency physicians missing hidden clues to a “can’t miss” diagnosis?

Image courtesy of EMS 12-Lead
A nine-year-old healthy girl at summer camp, running up a hill to chase her friend, collapses and fractures her left arm. She is seen in the local emergency department where she is diagnosed with a supracondylar fracture and dehydration. Three months later her brother is found dead in his bed in the morning.
A 26-year-old woman is asleep on the couch one week after the uneventful delivery of her first baby. The pizza delivery man rings the doorbell and when her husband arrives in the living room with the food he finds her gurgling and unresponsive. He initiates CPR and medics are able to restore sinus rhythm with defibrillation. She is diagnosed with seizure disorder at the local emergency department and started on Keppra. Two weeks later she has another episode shortly after her husband’s alarm wakes her from a sound sleep but this time she cannot be revived. Post mortem genetics reveal c-LQTS type 2 and her baby is also gene positive.
A 10-year-old boy is at swim team practice, a thunderstorm begins and a whistle blows to get the kids out of the pool. He jumps out quickly to a crash of thunder and has a witnessed face first syncopal episode that lasts seconds. He arrives in the ED cold and wet with a fractured nasal bone and lip laceration. He is told to eat more carbohydrates prior to practice. About six months later his father has cardiac arrest during a triathlon.
A 34-year-old lawyer is preparing for her third marathon by exercising daily at the gym. After a 45-minute workout she has a syncopal episode and falls off the treadmill, sustaining a forehead laceration but is otherwise uninjured. At the local ED, she is given the V.I.P. treatment since her law firm is a major malpractice litigator. The vitals are all normal except for a pulse of 42, just what is expected for this highly conditioned athlete. The plastic surgeon provides three sutures, the neurologist diagnoses “convulsive syncope,” is discharged. She returns 72 hours later in cardiac arrest but cannot be
revived.
What do all of these cases have in common? Each of these patients was eventually or retrospectively diagnosed with congenital long QT syndrome (c-LQTS). This should be distinguished from other causes of a prolonged QT interval, including electrolyte abnormalities or medications. The two may not be that distinct, however. Most authorities agree that those with torsades from acquired causes actually do have a concurrent underlying (but undiagnosed) congenital long QT syndrome as well. This article will focus on the congenital long QT syndrome, presentation, diagnosis and management of this sneaky, often obscured diagnosis that is more common than we think.
The incidence of the congenital long QT syndrome is about 1 in 2500 births. This is likely an underestimate as sudden death with a normal autopsy may be the first and only presentation. There are 12 genes responsible for 3 main types of c-LQTS. The presentation is one of aborted sudden death or brief arrhythmia. This usually manifests as sudden, without warning syncope, near syncope or prolonged syncope with seizures. Many are misdiagnosed with seizure disorder or vasovagal syncope.
The diagnosis is typically missed by at least one or more providers and generally takes an experienced electrophysiologist. If the 12-lead EKG demonstrates marked QT prolongation (>500ms) in the setting of a worrisome episode and family history, then the diagnosis is easy. Sometimes, the 12-lead EKG may be normal or near normal and only with provocative testing with a treadmill stress test or holter monitor will the diagnosis be made.
Is this diagnosis approached differently in children than adults? No, but the risk of sudden death varies with both age and gender. The highest risk tiers are those with QTc greater than 500 ms at any age and with any type of c-LQTS. Then, males with LQTS-1 and 2 are at highest risk prior to age 14. In females the risk increases with age with the highest risk age group after 14 and type 2.
Salient facts the EP must recognize
First, the history. Syncope in young people is an entirely different chief complaint than in older patients. The various “syncope rules” used for Grandma are mostly predicated on findings associated with left ventricular disease, especially coronary artery stenosis. Syncope in the young should have a different set of rules. And, to make it more complicated, most people will have benign vasovagal syncope at least once in their lifetime and patients with c-LQTS may have have both vasovagal syncope and syncope heralding sudden death.
It begins with the story. Syncope in long QT syndrome is essentially aborted torsades de pointes. It is generally without warning, abrupt and leaves the patient unable to slump to the ground without injury. There may be facial trauma or a fractured bone because there was no prodrome, no warning. Syncope in LQTS often leads to a seizure but by definition it is not the inciting event – torsades de pointes is. A family history of sudden death is common, as is a previous history of sudden unexplained syncope.
Fever is a trigger for LQTS type 2 and the first presentation may be a febrile “seizure” in a toddler. Startling noises, fright, and exertion are all LQTS torsades triggers and they vary in the different QT syndromes. Single vehicle car accidents, near drowning, syncope after starting new medications, SIDS and fainting while still lying in bed are all red flags. Age and gender are important as well. Congenital LQTS has two humps in its mortality curve — one is the school aged boy and the other is the female over age 18. The peri-partum timeframe is also particularly dangerous for the LQTS female.
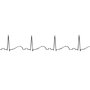
Next is the EKG. The QTc will be too long. How long is too long? And is it all just an absolute number? In general, a QTc less than 420 msec is likely normal. And a QTc greater than 500 msec is definitely abnormal and cause for referral. Most guidelines will say any QTc greater than 460 msec in a man and 470 msec in a female is abnormal. In the absence of significant variability in the R-R intervals and without any bundle pattern the end of the T wave complex should not be approaching the next P wave. Your eye will start to notice “too long” without needing to hand measure. The computer derived QTc should always be considered and should prompt careful inspection and measurement when reported as abnormal. Bradycardia may precipitate torsade in some forms of the syndrome and most LQTS EKGs have a distinct T wave morphology, bifid or notched.* Even with a borderline QTc the presence of notched T waves is highly concerning.
If LQTS is suspected on the basis of the history, family history and EKG, urgent cardiology consultation is appropriate. Specifically, an electrophysiologist will need to be involved. If the 12 lead EKG is borderline, than often a treadmill stress test will reveal subtle T wave abnormalities and a 24 hour holter can calculate QTc averages. Genetic testing will be positive in 75% of the cases but because all of the genes responsible for c-LQTS have not been discovered there are cases of the gene negative c-LQTS.
And once confirmed, how is this treated? The first step is often a beta-blocker such as nadolol or propranolol, titrated to heart rate blunting. There are three main types (although many others) of c-LQTS and type 1 responds most to beta-blocker therapy. Type 2 LQTS is treated with beta blockers and if ICD/pacer is present it responds well to active pacing as LQTS2 can be pause dependent. Type 3 patients are on a beta blocker and often mexilitine. The highest risk patients and those that have breakthrough events are considered for implantable cardioverter defibrillators and/or cervical denervation therapy. Avoidance of QT prolonging drugs, electrolyte abnormalities, dehydration, over-exertion, startling noises is recommended with all types of c-LQTS.
There are some conditions that have been long recognized as “can’t miss diagnoses” for the EP. Subarachnoid hemorrhage, acute myocardial infarction and bacterial meningitis are a few. With an untreated mortality approaching 25 percent, this eminently diagnosable and readily treatable condition should be added to the list.
Clues about Syncope
• Syncope in long QT syndrome is aborted sudden death
• Syncope in the young should be approached differently
• Use a careful approach to the 12 lead EKG in any child or adult with syncope looking specifically for QTc greater than 460 ms
• The clue to diagnosis is in the syncope story — sudden, without warning associated with facial trauma is a worrisome story for aborted sudden death.
• EKG in congenital LQTS — it’s not just the QTc; look for notched or bifid T waves
• Family history of sudden death, drowning, SIDS, single vehicle car accidents should be ascertained
• If long QT syndrome is suspected, cardiology consultation in ED is advised and often beta blocker therapy will be initiated
• Patients with QTc greater than 500 ms on their 12 lead EKG in ED should be seen by cardiology before discharge
Teaching Points for Patients
• Passing out can be a warning sign for a more serious condition and you will need to see a specialist to sort this out.
• Your EKG does not demonstrate a heart attack but is abnormal and could lead to arrhythmia.
• Long QT syndrome can be suspected from the ED but you need to see an experienced rhythm specialist to confirm the diagnosis
• Just to be extra cautious you should avoid all medications that could lengthen the QT interval, avoid dehydration, fever, startling noises
My LQTS Story

My friend next to me at boot camp class thought I had slumped over to tie my shoe but really I had a brief feeling of impending doom prior to a sudden, without warning syncopal episode. I was quite familiar with the prodrome of vasovagal syncope but this particular episode was different. I had a stress echo after that event which was read as normal and I was deemed incredibly fit and healthy. I decided to take a peek at my own exercise tracings and noticed that my ST segments were “odd” appearing, not quite ischemic, but not normal.
What I was looking at was that my T wave was slurring into my P wave during exertion. I then had a 12 lead resting EKG which revealed QT prolongation at 513 ms with notched and bifid T waves, classic for long QT type 2. I was started on nadolol 20 mg daily and it was titrated up to 40 mg, my running was limited to cardiac rehab and I began having severe symptomatic bradycardia on the nadolol. A pacemaker was recommended to tolerate the beta blocker. I went in for a routine pacemaker implant and just after induction of anesthesia had PVCs, followed by polymorphic VT and sustained torsades requiring defibrillation.
The next day, I went back for a dual chamber ICD. Shortly after, my genetics came back positive for LQTS type 2 and 2 of my four children are also positive. They are asymptomatic and followed closely on nadolol. We carry AEDs to the soccer fields and school trips and our kids are warned prior to any sudden alarms at school. We have embraced this frightening diagnosis and have chosen to live life with it.
Later that same year I saw an expert at the Mayo Clinic and was given the green light to start running again. I ran a personal best half marathon that fall, about 9 months after my ICD placement. I consider us the lucky ones. My goal is to teach my peers about this often concealed diagnosis that we have the chance to uncover and potentially save the lives of those at highest risk of sudden cardiac death.
Dr. Jennifer White practices emergency medicine at Doylestown Hospital.
7 Comments
Really found this article very interesting. I live in the UK and last year my 7 year old daughter, Violet had a sudden cardiac arrest whilst she was asleep. I heard her cough and went up and found her with no breath at all. My husband and I performed CPR for 7 minutes, that’s when the paramedics arrived at our house. By this time she was in full VF and was shocked with a defibrillator, CPR was then performed for a further 2 minutes and she came back to us. She had never been ill, never fainted and is so fit. After 3 weeks in hospital they diagnosed long qt, my husband and I, also our 13 year old daughter have all been tested and we don’t have it, also Violet’s genetic tests were negative as well. She had an ICD fitted and takes beta blockers. I know we are so blessed she survived and she is leaving life to the full, back ballroom dancing, swimming, playing netball and is so well. Her ICD has never gone off, but if it happens again she’s is protected.
My daughter Theodora Elise (8mo) is a Timothy Syndrome baby. I wish this type of silent killer didn’t lurk in people and love to see the brave stories of those who people who deal with it daily. I wish you and your family the all the best. Thank you for sharing with the world.
Great article but Fig. 1 is missing which should show: LQTS EKGs have a distinct T wave morphology, bifid or notched (Figure 1).
Thanks for sharing.
Since the image of a bifid T wave did not appear in the article I found an article which did and found that it was valuable in the context of the pediatric patient population – http://www.ijponline.net/content/pdf/1824-7288-35-17.pdf Otherwise the article was outstanding.
Jennifer, thanks for the article. My12 yo daughter was just diagnosed with lqts. Seems we all missed warning signs beginning with febrile seizure, continuing with compound arm fracture from a fall and 6 fainting episodes which were contributed to anxiety or vasovagil response. I hope with treatment she can live a normal life. It’s a scary diagnosis.
Thank you for writing the article and sharing your story. I am also a medical doctor living in Norway. I was diagnosed with LQTS type 1 about a year ago. Like you I kind of diagnosed my self, this would never have been picked up if it was not for me putting two and two together. That scares me and make it important to spread knowledge about LQTS as you do! I was started om betablockers and my four children were also tested. The result came back positive for three of them, so it turns out both my son aged 4 and my daughters ages 9 and 12 have LQTS. Last Monday I had my first episode of syncope while I was walking outside. No warning, fell on to the ground with my face first resulting considerable damage. So now I am in the hospital waiting to get considered for ICD. Thanks again for your work.
Twenty-seven years ago my heart stopped. Was taking two meds on the “No” list. Don’t know if that was the reason. The paramedics had to shock me several times and then the hospital had to do the same. They did not know what wrong and sent me to a bigger hospital. Saw several doctors who did not know what was wrong. Finally, one did and I got an ICD and Beta Blocker. I am eighty-three and have had only two shocks and that was because of old leads. Got a new ICD in my shoulder! I worry a lot about the drugs I should not have. Have to really watch the doctors when they give me medicine. I would like to know what you thinks about the drugs they give for Covid-19, also the drugs in the vaccines. My sister, died last year, had QT. We have been in touch with you since I found I had it. We learned so much from your brochures, more than from the doctors. Thank you so much for making it easier for us. I live in Ft. Payne, AL.